
Bs Phan Diễm Đoan Ngọc
Dịch từ: Fetology – Diagnosis and Management of the Fetal Patient, Third Edition
- Thể chai là một con đường chính kết nối hai bán cầu não. Đến tuần thứ 17 của thai kỳ, thể chai trưởng thành được hình thành.
- Khi bị bất sản thể chai(ACC), các sợi dây thần kinh không cắt ngang đường giữa mà tạo thành các bó sợi dày gọi là bó Probst, hướng ra phía sau dọc theo thành giữa của não thất bên. Các bó này thụt vào và tách các sừng trước của não thất bên.
- ACC có thể đơn độc, nhưng thường kết hợp với các dị tật và hội chứng di truyền khác.
- ACC xảy ra ở <1% dân số nói chung và từ 2% đến 3% dân số bị chậm phát triển.
- Việc phát hiện dãn não thất khi khám siêu âm định kỳ trước khi sinh nên được khảo sát kỹ hơn bằng siêu âm hình thái học để xác định sự hiện diện của thể chai
- Phát hiện nhất quán và dễ xác định nhất là cấu trúc hình giọt nước của não thất bên.
- Hình ảnh cộng hưởng từ trước khi sinh (MRI) và khám siêu âm 3D có thể hữu ích trong việc xác nhận chẩn đoán. MRI cũng có thể hữu ích trong việc xác định các bất thường tinh vi khác của não có thể không rõ ràng trên siêu âm nhưng có thể có tác động đến tiên lượng lâu dài.
- Diễn tiến tự nhiên của ACC được phát hiện trước sinh không được biết rõ do có ít trường hợp được báo cáo.
- Karyotype của thai nhi nên được khảo sát khi ACC được chẩn đoán trước sinh do có liên quan đến bất thường nhiễm sắc thể.
- Bệnh nhân được chẩn đoán ACC trước khi sinh có thể được quản lý theo các hướng dẫn chăm sóc trước khi sinh thường quy.
- Trẻ sơ sinh nghi ngờ mắc ACC nên được kiểm tra cẩn thận. MRI là lựa chọn để kiểm tra
- ACC cùng với các bất thường khác thường liên quan đến kết cục thần kinh kém. Kết cục lâu dài của ACC đơn độc vẫn chưa được làm sáng tỏ.
- Nguy cơ tái phát của ACC phụ thuộc vào nguyên nhân nền.
TỔNG QUÁT
Thể chai là con đường chính kết nối hai bán cầu não. Sự phát triển của thể chai bắt đầu trong tuần thứ 5 của bào thai với sự hình thành của các lớp đệm nguyên thủy (primitive lamina terminalis), dày lên để tạo thành tấm đệm (commissural plate). Các tế bào thần kinh đệm liên kết lại với nhau để tạo thành một cấu trúc giống như cầu nối, đóng vai trò dẫn đường cho các sợi thể đệm băng qua nếp gấp não dọc đến các mục tiêu của chúng ở phía cạnh bên của não (Rakic và Yakovlev, 1968). Thể chai trưởng thành được hình thành vào tuần thứ 17 của thai kỳ. Trong bất sản thể chai(ACC), các sợi tấm đệm không vượt qua đường giữa; thay vào đó, chúng tạo thành các bó sợi dày, được gọi là “bó Probst”, hướng ra phía sau dọc theo các vách giữa của não thất bên. Các bó này thụt vào và tách các sừng trước của não thất bên.
ACC có thể là một phát hiện riêng biệt; tuy nhiên, nó thường liên quan đến các dị tật và hội chứng di truyền khác bao gồm các sai lệch nhiễm sắc thể và bất thường chuyển hoá (Parrish và cộng sự, 1979; Jeret và cộng sự, 1987; Dobyns, 1989). Các bất thường liên quan đến hệ thần kinh trung ương (CNS) bao gồm dị dạng Chiari, dị tật di chuyển tế bào thần kinh bao gồm lissencephaly (nhẵn não hay não trơn/não mịn/não phẳng), schizencephaly (nứt não), pachygyria (hồi não rộng) và polymicrogyria, encephaloceles (thoát vị não), dị tật Dandy– Walker, holoprosencephaly và olivoponto – thoái hóa tiểu não (Barkovich và Norman, 1988). Dị tật ngoại sọ bao gồm các bất thường của khuôn mặt và hệ thống tim mạch, sinh dục, tiêu hóa, hô hấp và cơ xương (Parrish và cộng sự, 1979; Franco và cộng sự, 1993; Kozlowski và Ouvrier, 1993).
TỶ LỆ MẮC BỆNH
ACC ước tính xảy ra ở 0,3% đến 0,7% dân số thế giới và 2% đến 3% dân số chậm phát triển (Freytag và Lindenberg, 1967; Grogono, 1968; Jeret và cộng sự, 1986). Trong một loạt lớn bệnh nhân được chẩn đoán mắc bệnh chuyển hóa, 17% cho thấy có sự bất thường của thể chai bằng chụp cắt lớp vi tính (CT), siêu âm hoặc khám nghiệm tử thi (Bamforth và cộng sự, 1988).
Trong một loạt 4122 mẫu mổ tử thi chu sinh hoặc sơ sinh, tỷ lệ dị tật thần kinh trung ương là 8,8%, và trong số 363 dị tật thần kinh trung ương được chẩn đoán, ACC chiếm 4,1% (Pinar và cộng sự, 1998)
HÌNH ẢNH SIÊU ÂM
Thông thường, chỉ những mặt cắt đứng dọc giữa (mid sagittal) hoặc đứng ngang giữa (midcoronal) mới cho được hình ảnh rõ ràng về thể chai. Những mặt cắt như vậy có thể đạt được khi kiểm tra siêu âm qua ổ bụng tiêu chuẩn của hầu hết các thai nhi ở tư thế ngôi mông hoặc ngôi ngang. Đối với những thai nhi ở ngôi đầu, siêu âm qua âm đạo là kỹ thuật ưu tiên.
Trên mặt cắt đứng dọc sagittal, thể chai xuất hiện như một dải siêu âm, được phân giới trên và dưới bởi hai đường hồi âm. Đường phía trên phát sinh từ per- icallosal cistern; đường phía dưới bắt nguồn từ fornix và trần của vách trong suốt (cavum pellucidum). Hình ảnh siêu âm Doppler màu có thể chứng minh pericallosal artery, một nhánh của động mạch não trước quét theo hình tròn trên tiểu thể. Trên mặt cắt đứng ngang, thể chai dường như tạo thành trần của vách trong suốt và sừng trán.
Các dấu hiệu thần kinh cổ điển để chẩn đoán ACC đã được mô tả bởi Davidoff và Dyke (1934). Tiêu chí của họ đã được áp dụng cho chẩn đoán CT và cũng có thể được áp dụng cho siêu âm (Skidmore và cộng sự, 1983). Các phát hiện bao gồm sự vắng mặt của thể chai và vách trong suốt và một loạt các phát hiện gián tiếp, bao gồm
- tăng sự tách biệt của sừng trước và thân của não thất bên;
- sự giãn ra tương đối của sừng chẩm của các não thất bên;
- đường viền lõm giữa lõm của não thất bên do sự phát triển của các bó Probst;
- sự giãn nở chung và sự dịch chuyển lên trên của não thất ba;
- định hướng bên bất thường của nếp gấp não giữa(Comstock và cộng sự, 1985; Bertino và cộng sự, 1988; Sandri và cộng sự, 1988; Pilu và cộng sự, 1993; Maheut-Lourmiere và Paillet, 1998).
Trong một loạt 141 trường hợp dãn não thất, ACC được ghi nhận trong 16 trường hợp (11%) (Valat và cộng sự, 1998). Trong một loạt 82 trường hợp khác của chứng dãn não thất nhẹ, có 7 trường hợp (9%) là ACC (Vergani và cộng sự, 1998). Do đó, việc phát hiện thấy dãn não thất nhẹ trong quá trình siêu âm định kỳ trước khi sinh nên thúc đẩy một cuộc tìm kiếm có chủ đích để xác nhận sự hiện diện của thể chai.
Chẩn đoán bằng siêu âm của ACC trước sinh khó khăn hơn là sau sinh. Chẩn đoán trước sinh sớm nhất được thực hiện khi thai được 19 tuần (Pilu và cộng sự, 1993). Bởi vì thể chai không được hình thành bình thường cho đến 18 tuần, hầu hết các trường hợp được báo cáo đã được chẩn đoán trong tam cá nguyệt thứ ba (Bertino và cộng sự, 1988; Bennett và cộng sự, 1996). Khó khăn này trong chẩn đoán trước khi sinh thường là do các bất thường não thất đặc trưng có thể khá tinh tế trên các mặt cắt ngang là mặt cắt thường được lấy nhất. Tốt hơn là sử dụng các mặt cắt đứng ngang (coronal) và đứng dọc (sagittal) để chẩn đoán các bất thường đặc trưng của não thất; tuy nhiên, những mặt cắt này không được sử dụng phổ biến trong chẩn đoán tiền thai.
Siêu âm thường quy não thai nhi thường được thực hiện với các mặt cắt ngang không cho phép hình dung được thể chai. Tuy nhiên, việc dãn ngã ba não thất của não thất bên được đánh giá cao với những mặt cắt này. Trong loạt ACC được chẩn đoán trước sinh lớn nhất, 34 trong số 35 thai nhi có số đo não thất bên 10 mm (Pilu và cộng sự, 1993). Khi đã quan sát thấy dãn não thất bên, các phát hiện siêu âm khác có thể tìm thấy ACC. Phát hiện nhất quán và dễ xác định nhất là cấu trúc hình giọt nước của não thất bên (Hình 6-1). Trong mặt cắt ngang, dãn ngã ba não thất và sừng chẩm và sự tách các cơ quan ra để tạo ra một hình dạng dễ nhận biết. Cấu hình giọt nước của não thất chưa được ghi nhận trong các điều kiện khác ngoài ACC, và nó được cho là một dấu hiệu cụ thể để chẩn đoán chứng bất sản thể chai(Pilu và cộng sự, 1993). Để ghi nhận trực tiếp tổn thương thể chai, điều quan trọng là phải cắt được mặt cắt đứng dọc giữa (midsagittal) và đứng ngang giữa (midcoronal) khi phát hiện dãn não thất hoặc não thất hình giọt nước.
Chẩn đoán trước sinh về ACC bán phần đã được báo cáo (Hình 6-2) (Lockwood và cộng sự, 1988; Pilu và cộng sự, 1993). Lịch sử tự nhiên của ACC bán phần là không chắc chắn, và các phát hiện não liên quan đến nó có lẽ tinh vi hơn so với dạng hoàn chỉnh. Chẩn đoán trước sinh sẽ không thể thực hiện được trong nhiều trường hợp. Vách trong suốt không được xác định trong các trường hợp ACC hoàn toàn nhưng có thể được hình dung trong các trường hợp ACC một phần. Đánh giá cấu trúc này bằng siêu âm có khả năng hữu ích trong việc kiểm tra định kỳ, thông báo cho bác sĩ chuyên khoa chẩn đoán để thực hiện thêm các mặt cắt đứng ngang và đứng dọc để quan sát trực tiếp thể chai. Sự mở rộng của khe liên bán cầu và sự dịch chuyển lên trên của não thất ba có thể được ghi nhận trong khoảng một nửa số trường hợp trước khi sinh. Sự sắp xếp lệch tâm của hồi não giữa chỉ được thấy trên siêu âm ở tam cá nguyệt thứ ba. Điều này có lẽ là do bề mặt của các bán cầu nhẵn trong tam cá nguyệt thứ hai, với các hồi não thứ phát chỉ xuất hiện khi bắt đầu tam cá nguyệt thứ ba. Trong nhiều trường hợp ACC, siêu âm trước khi sinh có thể chỉ khiến người ta nghi ngờ chẩn đoán, trong khi cần chụp cộng hưởng từ trước khi sinh (MRI) để xác định chính xác chẩn đoán (Glenn và cộng sự, 2005). Trong một loạt 14 trường hợp ACC, siêu âm chỉ xác nhận chẩn đoán trong 4 trường hợp, trong khi MRI xác nhận chẩn đoán trong 13 trường hợp (d’Ercole và cộng sự, 1998). Trong một loạt 20 trường hợp ACC khác, MRI trước sinh cho chẩn đoán dương tính trong 19 trường hợp (Brisse và cộng sự, 1998). Trong một loạt gần đây gồm 10 trường hợp nghi ngờ có bất thường về thể chai trên siêu âm, 8 trường hợp được xác nhận qua MRI và 2 trường hợp được phát hiện có thể chai bình thường (Glenn và cộng sự, 2005). MRI cũng hữu ích trong việc xác định các bất thường bổ sung của não mà siêu âm không thấy rõ, chẳng hạn như bất thường về sự hình thành rãnh não. 63% trường hợp (5/8) có bất thường thể chai được xác nhận trên cả siêu âm và MRI được phát hiện có thêm bất thường trên MRI. Những phát hiện thêm vào này đã được xác nhận trên MRI hoặc khám nghiệm tử thi ở ba trong số năm bệnh nhân. Thông tin thu được từ MRI là rất có giá trị cho việc tư vấn cho bệnh nhân vì các bất thường về não có liên quan đến kết cục xấu về lâu dài. Ngoài ra, siêu âm ba 3D cũng có thể hữu ích để xác nhận chẩn đoán (Kalache và cộng sự, 2006; Pilu và cộng sự, 2006).


CHẨN ĐOÁN PHÂN BIỆT
Sự giãn của ngã ba não thất và sừng chẩm thường là phát hiện siêu âm nổi bật nhất; do đó, bất cứ nguyên nhân nào gây ra não úng thủy cũng nằm trong chẩn đoán phân biệt đối với ACC (xem Chương 16). Trong ACC, sừng chẩm thường to hơn, trong tương quan với hệ thống não thất còn lại, hơn là khi quan sát thấy ở các dạng não úng thủy khác. Khi não thất ba bị giãn, chẩn đoán phân biệt bao gồm các nang ở đường giữa khác như vách trong suốt (cavum septum pellucidum), cavum vergae, interhemispheric arachnoid cyst, and medial porencephalic cyst
ACC có liên quan đến nhiều hội chứng di truyền dễ nhận biết (xem Bảng 6-1) (Bamforth và cộng sự, 1988; Paul và cộng sự, 2007). ACC cũng có liên quan đến các bất thường của nhiễm sắc thể 8, 13 và 18 (Jeret và cộng sự, 1987). ACC cũng có thể liên quan đến một số bệnh lý bẩm sinh của quá trình chuyển hóa, bao gồm tăng đường huyết không keton, hội chứng Zellweger, thiếu hụt adenylosuccinase, thiếu pyruvate dehydrogenase, loạn dưỡng tăng sinh ở trẻ sơ sinh neonatal adrenoleukodystrophy), bệnh Menkes, hội chứng Leigh và axit glutaric niệu loại II (Dobyns, 1989; Kolodny , 1989; Blum và cộng sự, 1990). Nhiều tác nhân gây dị tật khác cũng có thể được coi là nguyên nhân có thể gây ra ACC, bao gồm rượu, valproate, cocaine, rubella và vi rút cúm (Bảng 6-1) (Friedman và Cohen, 1947; Cornover và Roessmann, 1990; Dominguez và cộng sự. , 1991; Lindhout và cộng sự, 1992).

DIỄN TIỄN TỰ NHIÊN TRƯỚC SINH
Người ta biết rất ít về diễn tiến tự nhiên của ACC khi nó xảy ra như một phát hiện riêng biệt vì có tương đối ít trường hợp được báo cáo. Trong một loạt 37 trường hợp mang thai với ACC được chẩn đoán trước, 28 trường hợp dẫn đến việc chấm dứt thai kỳ có chọn lọc, và 9 trường hợp còn lại là tiếp tục thai kỳ và sinh trẻ sống(Hatem-Gantzer và cộng sự, 1998). Bốn trong số 9 trẻ sơ sinh này đã chết trong giai đoạn sau khi sinh. Có vẻ như sự hiện diện của ACC đơn độc sẽ không thay đổi chăm sóc tiền sinh
QUẢN LÝ MANG THAI
Việc chẩn đoán ACC đòi hỏi phải khám xét cẩn thận giải phẫu của thai nhi để tìm các bất thường khác trong sọ và ngoài sọ. Mối liên quan giữa ACC và các bất thường về nhiễm sắc thể đã được Serur et al. (1988), sau khi xem xét 100 trường hợp. MRI có giá trị để xác nhận chẩn đoán và loại trừ dị tật nội sọ nào khác cùng tồn tại (Brisse và cộng sự, 1998; d’Ercole và cộng sự, 1998; Glenn và cộng sự, 2005). Siêu âm ba chiều cũng có thể hữu ích (Kalache và cộng sự, 2006; Pilu và cộng sự, 2006).
Trong báo cáo của Serur và cộng sự, trisomy 18 thể toàn phần hay thể khảm được tìm thấy trong 29 trường hợp, trisomy 8 trong 21 trường hợp, trisomy 13 trong 20, và một loạt các tình trạng khác trong các trường hợp còn lại. Do đó, người ta công nhận rằng các nhiễm sắc thể 8, 13 và 18 có ảnh hưởng trực tiếp đến sự phát triển của thể chai. Trong đánh giá của Pilu và cộng sự (1993) về chẩn đoán ACC ở thai nhi, ba thai nhi có bất thường nhiễm sắc thể số 8 có ACC là phát hiện siêu âm duy nhất. Do đó, thông lệ của chúng tôi là tiến hành chọc ối để khảo sát nhiễm sắc thể thai nhi một cách độc lập với sự hiện diện các bất thường trên siêu âm.
Nếu một ACC đơn độc được phát hiện và các nhiễm sắc thể bình thường, không có chỉ định thay đổi chăm sóc sản khoa thường quy. Chẩn đoán này không cần thiết phải sinh tại bệnh viện chăm sóc cấp cao. Nên tham khảo thêm ý kiến của các chuyên gia di truyền học và thần kinh nhi khoa. Tuy nhiên, sự hiện diện của các bất thường cấu trúc liên quan khác cần phải nhanh chóng chuyển đến các bác sĩ chuyên khoa nhi thích hợp, và trong tình huống này, bệnh nhân nên được chuyển đến bệnh viện chăm sóc cấp cao.
Sinh qua ngã âm đạo được khuyến cáo trừ khi có não úng thủy đáng kể kèm theo tật đầu to. Không nên khởi phát chuyển dạ trước kỳ hạn trừ khi có chỉ định sản khoa.
CAN THIỆP BÀO THAI
Không có khuyến cáo can thiệp bào thai cho vấn đề này.
ĐIỀU TRỊ TRẺ SƠ SINH
Cần phải kiểm tra chi tiết trẻ sơ sinh. MRI nên được chỉ định do có thể phát hiện các bất thường tinh vi mà phương pháp khác bỏ sót, gồm cả CT và siêu âm (Davidson và cộng sự, 1985; Barkovich và Norman, 1988). Có thể cần thực hiện thêm các xét nghiệm chuyển hóa nếu nghi ngờ có bất thường bẩm sinh về chuyển hóa.
ĐIỀU TRỊ PHẪU THUẬT
Không có chỉ định phẫu thuật đối với ACC đơn độc; tuy nhiên, phẫu thuật có thể được chỉ định cho thêm các bệnh lý nội sọ hoặc ngoại sọ. Một số trường hợp có thể cần đặt shunt não thất – màng bụng để điều trị chứng giãn não thất tiến triển và tăng kích thước đầy(Lacey, 1985).
KẾT CỤC DÀI HẠN
Trẻ em bị ACC và nhiều dị tật bẩm sinh nặng có nguy cơ cao bị chậm phát triển thần kinh, đặc biệt nếu có dị tật sọ mặt. Sự hiện diện của co giật ở trẻ sơ sinh hoặc trẻ nhỏ là một dấu hiệu tiên lượng đáng ngại; hầu hết những đứa trẻ này chậm phát triển nghiêm trọng hoặc có biểu hiện chậm phát triển (Lacey, 1985). Trẻ có thêm các bất thường trong não cũng có vẻ có tiên lượng xấu, trẻ đa phần chậm phát triển. Chẩn đoán ACC nên cảnh báo cho bác sĩ về khả năng chậm phát triển thần kinh nghiêm trọng và các cơn co giật sau đó.
Hiện tại, không có số liệu cụ thể nào về nguy cơ cho các bậc cha mẹ liên quan đến sự phát triển thần kinh ở trẻ em bị ACC đơn độc. Vergani và cộng sự. (1988) báo cáo rằng ba trẻ sơ sinh được chẩn đoán trước khi sinh có ACC có chỉ số thông minh bình thường hoặc mức ngưỡng khi khám theo dõi 3 năm. Trong loạt bài của Pilu et al. (1993), số trẻ phát triển bình thường được tìm thấy ở 9 trong số 11 trường hợp không cùng huyết thống với thời gian theo dõi trong khoảng từ 6 tháng đến 11 năm, và phát triển mức ngưỡng được tìm thấy trong hai trường hợp còn lại. Sự phát triển thần kinh của trẻ sơ sinh sống sót được đánh giá bằng bài kiểm tra Brunet-Lezine và Thang đo trí tuệ Stanford – Binet. Ngay cả khi có trí thông minh bình thường, ACC có thể liên quan đến những khiếm khuyết về nhận thức tinh tế (Temple et al., 1989, 1990; Jeeves, 1991). Mối quan hệ có thể xảy ra giữa ACC và rối loạn tâm thần cũng đã được đề xuất (Swayze và cộng sự, 1990).
Trong một cuộc khảo sát của ACC từ Vương quốc Anh, hai phần ba trong số 59 bệnh nhân bị ảnh hưởng bị động kinh, một nửa số trường hợp trưởng thành bị suy giảm trí tuệ và một phần ba bị rối loạn tâm thần (Taylor và David, 1998). Tuy nhiên, loạt bài này và loạt bài khác có thể bị sai lệch do chưa được ghi nhận đầy đủ các trường hợp ACC không triệu chứng. Trong một loạt 10 trẻ khác được chẩn đoán ACC trước khi phẫu thuật, tất cả đều có kết quả phát triển bình thường sau 3 năm theo dõi, mặc dù co giật do sốt được báo cáo là xảy ra thường xuyên hơn dự kiến (Moutard và cộng sự, 1998).
Goodyear và cộng sự. (2001) so sánh bệnh nhân mắc ACC được chẩn đoán trước sinh(14) với những bệnh nhân được chẩn đoán ở giai đoạn sau khi sinh (61). Các bệnh nhân được đánh giá về các mốc phát triển cũng như các vấn đề lâm sàng khác. ACC phổ biến hơn ở nam giới hơn nữ giới, và ACC bán phần phổ biến hơn ở nhóm được chẩn đoán sau sinh. ACC toàn phần có tiên lượng xấu hơn so với ACC bán phần, đặc biệt khi nó kết hợp với các bất thường khác, nhất là là các bất thường của thần kinh trung ương. Chỉ những bệnh nhân bình thường về phát triển thần kinh mới ở trong nhóm ACC bán phần đơn độc. Các kết quả bất lợi về thần kinh bao gồm co giật, chậm phát triển thể chất và chậm phát triển trí tuệ, cũng như cần được giúp đỡ đặc biệt ở trường.
Gần đây hơn, Francesco et al. (2006) đánh giá 9 trẻ từ 2 đến 16 tuổi đã được chẩn đoán mắc chứng ACC trong thời kỳ trước khi sinh. Chẩn đoán đã được xác nhận trong giai đoạn sau sinh. Sáu đứa trẻ đã bị ACC đơn độc, ba trong số chúng bị nhược cơ toàn thân, trong khi ba đứa khác có phát triển thần kinh bình thường. Ba trẻ với những phát hiện kết hợp khác không có kết cục thần kinh bình thường. Các tác giả kết luận rằng nếu ACC kết hợp với các dị tật khác thì có khả năng kết cục xấu. Tuy nhiên, nếu không, sẽ khó khăn khi dự đoán liệu có kết cục thần kinh bình thường hay không.
Liên quan đến ACC bán phần, Volpe et al. (2006) gần đây đã đánh giá 19 trường hợp được chẩn đoán trước sinh. Mười trường hợp xảy ra liên quan đến các dị thường khác. Chín trường hợp ban đầu được cho là đơn độc. Một trong số chín trường hợp được phát hiện có thêm dị tật não sau khi chụp MRI, và trường hợp này đã bị mất dấu theo dõi. Có 11 ca đẻ sống, 6 ca bỏ thai và 2 ca tử vong chu sinh. Ba mươi hai phần trăm (6/19) trường hợp được theo dõi tâm lý vận động bình thường. Tất cả các trường hợp này đều có ACC bán phần đơn độc. Hai trường hợp ACC bán phần đơn độc có kết cục kém thuận lợi hơn, một trong số đó có chậm phát triển đáng kể và giảm trương lực cơ. Tương tự như các trường hợp ACC toàn phần đơn độc, ACC bán phần đơn độc có thể liên quan đến các kết cục thần kinh bất lợi đáng kể.
NGUY CƠ DI TRUYỀN VÀ TÁI PHÁT
Nguy cơ tái phát của ACC, cho dù nó đơn độc hay có liên quan bẩm sinh về chuyển hóa hoặc hội chứng di truyền, phụ thuộc vào nguyên nhân cơ bản. Nếu ACC liên quan đến thể lệch bội, nguy cơ tái phát là 1% hoặc nguy cơ lệch bội liên quan đến tuổi mẹ, tùy theo giá trị nào lớn hơn. Trường hợp ACC đơn độc mà không rõ nguyên nhân, nguy cơ tái phát có thể vào khoảng từ 2% đến 3% (Young và cộng sự, 1985). ACC là một tiêu chí được biết đến để chẩn đoán một số hội chứng, chẳng hạn như hội chứng Aicardi, Andermann và acrocallosal syndrome(Philip et al., 1998). Trong khi ACC thường được coi là tự phát, một số trường hợp có tính chất gia đình đã được báo cáo (Naritomi và cộng sự, 1997).
Cùng chuyên mục
-
SỬ DỤNG MOLNUPIRAVIR TRONG ĐỘ TUỔI SINH SẢN
SỬ DỤNG MOLNUPIRAVIR TRONG ĐỘ TUỔI SINH SẢN - NHỮNG…
-

CHO CON BÚ KHI BẠN ĐANG MANG THAI
CHO CON BÚ KHI BẠN ĐANG MANG THAI – ĐƯỢC…
-

HỘI CHỨNG HẬU COVID 19
Hiện nay, nhiều bầu liên hệ bác sĩ vì sau…
-
TIÊM NGỪA COVID 19 CHO TRẺ 5-11 TUỔI
NHỮNG ĐIỀU BẠN CẦN BIẾT KHI TIÊM NGỪA COVID 19…
-
SẨY THAI SỚM
SẨY THAI SỚM LÀ GÌ? Là mất thai trước 13…
-
Lộ tuyến cổ tử cung
Lộ tuyến cổ tử cung là gì? Có gây ung…
-
DỊ TẬT DANDY–WALKER VÀ BIẾN THỂ
BS Phan Diễm Đoan Ngọc Dị tật Dandy – Walker…
-
THAI VÔ SỌ (ANENCEPHALY)
Bs Phan Diễm Đoan Ngọc Dịch từ: Fetology - Diagnosis…
-
BẠN LÀ F0, CÓ ẢNH HƯỞNG ĐẾN EM BÉ KHÔNG?
[embed]https://www.youtube.com/watch?v=Adot9b8WSwg[/embed]
-

TRỮ LẠNH NOÃN là gì?
Trữ lạnh noãn là gì? Phụ nữ sanh con muộn…
-

Thai chậm tăng trưởng trong tử cung
Bs Phan Diễm Đoan Ngọc Một ngày bạn đi khám…
-

SONG THAI
BS PHAN DIỄM ĐOAN NGỌC Mang trong mình không phải…
-

QUAN HỆ TÌNH DỤC KHI MANG THAI
BS Phan Diễm Đoan Ngọc Bạn mến, quan hệ tình…
-
VỆ SINH ÂM ĐẠO TRONG THAI KỲ
BS. Phan Diễm Đoan Ngọc Vài điều về sinh lý…
-

XÉT NGHIỆM NIPT( Sàng lọc lệch bội nhiễm sắc thể…
BS PHAN DIỄM ĐOAN NGỌC Khi mang thai, hẳn…
-

TÁO BÓN VÀ BỆNH TRĨ
BS PHAN DIỄM ĐOAN NGỌC Hễ có bạn nào đến…
-

CHUỘT RÚT (VỌP BẺ) KHI MANG THAI
BS PHAN DIỄM ĐOAN NGỌC - BV TỪ DŨ Chuột…
-

ĐAU ĐẺ VÀ CÁC PHƯƠNG PHÁP GIẢM ĐAU
Khi gần đến ngày khai hoa nở nhụy, bên cạnh…
-
LỰA CHỌN SANH THƯỜNG HAY SANH MỔ?
BS Phạm Thanh Hoàng Nhiều sản phụ khi gần đến…
-

Khi nào phải tới bệnh viện?
Bs Phan Diễm Đoan Ngọc - BV Từ Dũ Khi…
-

PHẢI LÀM GÌ KHI QUÊN UỐNG VIÊN THUỐC TRÁNH THAI?
BSCK1. Nguyễn Phương Nam - Bệnh viện Hùng Vương. Hiện…
-

Viêm nướu răng trong thai kỳ
Hôm rồi khám đến 2 chị bầu bị viêm nướu.…
-

ĐỪNG TỰ Ý NGƯNG THUỐC HEN SUYỄN KHI CÓ THAI
ĐỪNG TỰ Ý NGƯNG THUỐC HEN SUYỄN KHI CÓ THAI!!!…
-

PHẢI LÀM GÌ KHI BÀ BẦU BỊ CHÓ CẮN?
BS. Trần Thị Minh Châu. Một chị bầu người quen…
-
THỦY ĐẬU KHI MANG THAI
BỊ THỦY ĐẬU KHI ĐANG CÓ THAI – BẠN PHẢI…
-
SIÊU ÂM THAI CÓ HẠI GÌ KHÔNG?
BS Phan Diễm Đoan Ngọc Hôm rồi có một bạn…
-
LẦN TRƯỚC SANH MỔ, LẦN NÀY CÓ SANH THƯỜNG ĐƯỢC…
BS. Phạm Thanh Hoàng Đây là 1 câu hỏi rất…
-

MÃN KINH
BS PHAN DIỄM ĐOAN NGỌC - BV TỪ DŨ Chắc…
-
BĂNG HUYẾT SAU SANH
BS ĐOÀN TRUNG HIẾU - BV TỪ DŨ Thỉnh thoảng…
-

VIÊM ÂM ĐẠO
BS. Phạm Thanh Hoàng VIÊM ÂM ĐẠO LÀ GÌ? CÓ…
-

ĐAU LƯNG TRONG THAI KỲ
BS Đoàn Trung Hiếu - BV Từ Dũ ✅ NGUYÊN NHÂN…
-
THAI LƯU SỚM
BS PHAN DIỄM ĐOAN NGỌC - BV TỪ DŨ Hồi…
-
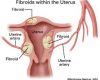
NHÂN XƠ TỬ CUNG
BS.Phạm Thanh Hoàng- Giảng viên ĐHYD, BV Từ Dũ NHÂN…
-
SANH GIÚP
BS Đoàn Trung Hiếu - BV Từ Dũ Bài viết…
-

Nhiễm trùng vết mổ lấy thai
BS Nguyễn Phương Nam - BV Hùng Vương Nhân dịp…
-

NGHÉN (THAI HÀNH)
THS.BS.Phan Diễm Đoan Ngọc NGHÉN là tình trạng rất phổ…
-

ĐẾM CỬ ĐỘNG THAI
THS.BS. Phan Diễm Đoan Ngọc CỬ ĐỘNG THAI LÀ GÌ?…
-

MANG THAI VÀ CHUYỆN TIÊM NGỪA
BSCK1. Nguyễn Phương Nam – Bệnh viện Hùng Vương. Phụ…
-

THAI NHỎ, NHẸ CÂN – NỖI LO CỦA MẸ
BS CK1. NGUYỄN PHƯƠNG NAM – BỆNH VIỆN HÙNG VƯƠNG.…
-

TIỂU ĐƯỜNG (ĐÁI THÁO ĐƯỜNG) THAI KỲ
Bs. Phạm Thanh Hoàng Giảng viên đại học Y Dược…
-

SANH KHÔNG ĐAU
BS. Phạm Thanh Hoàng Giảng viên ĐHYD TPHCM - BV…
-

CẤY QUE NGỪA THAI
ThS.BS. Phan Diễm Đoan Ngọc Chào bạn, chọn biện pháp…
-
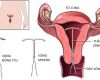
VÒNG TRÁNH THAI
VÒNG TRÁNH THAI (DỤNG CỤ TỬ CUNG) ThS.BS. Phan…
-
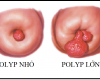
BIẾN ĐỔI LÀNH TÍNH CỔ TỬ CUNG
BIẾN ĐỔI LÀNH TÍNH CỔ TỬ CUNG: LỘ TUYẾN CỔ…
-

MẤT NGỦ KHI MANG THAI
BS. TRẦN THỊ MINH CHÂU. Mất ngủ khi mang thai…
-

PHỤ NỮ NÊN ĂN GÌ ĐỂ DỄ CÓ THAI?
BS. TRẦN THỊ MINH CHÂU ❓Bạn có biết: Khả năng…
-

DÙNG MỸ PHẨM TRONG THAI KỲ
ThS.BS. Phan Diễm Đoan Ngọc Nhân hôm nay có một…
-

DU LỊCH, ĐI LẠI TRONG THAI KỲ
BS.Phạm Thanh Hoàng Sắp đến Tết, nhiều người sẽ lên…
-

CANXI TRONG THAI KỲ
THS.BS.Phan Diễm Đoan Ngọc - BV Từ Dũ Hẳn bà…
-

LỢI ÍCH CỦA DẦU CÁ OMEGA-3 TRONG THAI KỲ
THS.BS.Phan Diễm Đoan Ngọc Trong thai kỳ, ngoài việc bổ…
-

THUỐC HỖ TRỢ PHỔI LÀ GÌ?
BS CK1. Nguyễn Phương Nam – Bệnh viện Hùng Vương.…
-

NÊN TIÊM NGỪA GÌ TRƯỚC KHI KẾT HÔN ?
BS. TRẦN THỊ MINH CHÂU Câu hỏi: Chào Bác sĩ,…
Có liên quan khác
-
SỬ DỤNG MOLNUPIRAVIR TRONG ĐỘ TUỔI SINH SẢN
SỬ DỤNG MOLNUPIRAVIR TRONG ĐỘ TUỔI SINH SẢN - NHỮNG…
-
CHO CON BÚ KHI BẠN ĐANG MANG THAI
CHO CON BÚ KHI BẠN ĐANG MANG THAI – ĐƯỢC…
-
HỘI CHỨNG HẬU COVID 19
Hiện nay, nhiều bầu liên hệ bác sĩ vì sau…
-
TIÊM NGỪA COVID 19 CHO TRẺ 5-11 TUỔI
NHỮNG ĐIỀU BẠN CẦN BIẾT KHI TIÊM NGỪA COVID 19…
-
SẨY THAI SỚM
SẨY THAI SỚM LÀ GÌ? Là mất thai trước 13…
-
Lộ tuyến cổ tử cung
Lộ tuyến cổ tử cung là gì? Có gây ung…
-
DỊ TẬT DANDY–WALKER VÀ BIẾN THỂ
BS Phan Diễm Đoan Ngọc Dị tật Dandy – Walker…
-
THAI VÔ SỌ (ANENCEPHALY)
Bs Phan Diễm Đoan Ngọc Dịch từ: Fetology - Diagnosis…
-
BẠN LÀ F0, CÓ ẢNH HƯỞNG ĐẾN EM BÉ KHÔNG?
[embed]https://www.youtube.com/watch?v=Adot9b8WSwg[/embed]
-
TRỮ LẠNH NOÃN là gì?
Trữ lạnh noãn là gì? Phụ nữ sanh con muộn…
-
Thai chậm tăng trưởng trong tử cung
Bs Phan Diễm Đoan Ngọc Một ngày bạn đi khám…
-
SONG THAI
BS PHAN DIỄM ĐOAN NGỌC Mang trong mình không phải…
-
QUAN HỆ TÌNH DỤC KHI MANG THAI
BS Phan Diễm Đoan Ngọc Bạn mến, quan hệ tình…
-
VỆ SINH ÂM ĐẠO TRONG THAI KỲ
BS. Phan Diễm Đoan Ngọc Vài điều về sinh lý…
-
XÉT NGHIỆM NIPT( Sàng lọc lệch bội nhiễm sắc thể…
BS PHAN DIỄM ĐOAN NGỌC Khi mang thai, hẳn…
-
TÁO BÓN VÀ BỆNH TRĨ
BS PHAN DIỄM ĐOAN NGỌC Hễ có bạn nào đến…
-
CHUỘT RÚT (VỌP BẺ) KHI MANG THAI
BS PHAN DIỄM ĐOAN NGỌC - BV TỪ DŨ Chuột…
-
ĐAU ĐẺ VÀ CÁC PHƯƠNG PHÁP GIẢM ĐAU
Khi gần đến ngày khai hoa nở nhụy, bên cạnh…
-
LỰA CHỌN SANH THƯỜNG HAY SANH MỔ?
BS Phạm Thanh Hoàng Nhiều sản phụ khi gần đến…
-
Khi nào phải tới bệnh viện?
Bs Phan Diễm Đoan Ngọc - BV Từ Dũ Khi…
-
PHẢI LÀM GÌ KHI QUÊN UỐNG VIÊN THUỐC TRÁNH THAI?
BSCK1. Nguyễn Phương Nam - Bệnh viện Hùng Vương. Hiện…
-
Viêm nướu răng trong thai kỳ
Hôm rồi khám đến 2 chị bầu bị viêm nướu.…
-
ĐỪNG TỰ Ý NGƯNG THUỐC HEN SUYỄN KHI CÓ THAI
ĐỪNG TỰ Ý NGƯNG THUỐC HEN SUYỄN KHI CÓ THAI!!!…
-
PHẢI LÀM GÌ KHI BÀ BẦU BỊ CHÓ CẮN?
BS. Trần Thị Minh Châu. Một chị bầu người quen…
-
THỦY ĐẬU KHI MANG THAI
BỊ THỦY ĐẬU KHI ĐANG CÓ THAI – BẠN PHẢI…
-
SIÊU ÂM THAI CÓ HẠI GÌ KHÔNG?
BS Phan Diễm Đoan Ngọc Hôm rồi có một bạn…
-
LẦN TRƯỚC SANH MỔ, LẦN NÀY CÓ SANH THƯỜNG ĐƯỢC…
BS. Phạm Thanh Hoàng Đây là 1 câu hỏi rất…
-
MÃN KINH
BS PHAN DIỄM ĐOAN NGỌC - BV TỪ DŨ Chắc…
-
BĂNG HUYẾT SAU SANH
BS ĐOÀN TRUNG HIẾU - BV TỪ DŨ Thỉnh thoảng…
-
VIÊM ÂM ĐẠO
BS. Phạm Thanh Hoàng VIÊM ÂM ĐẠO LÀ GÌ? CÓ…
-
ĐAU LƯNG TRONG THAI KỲ
BS Đoàn Trung Hiếu - BV Từ Dũ ✅ NGUYÊN NHÂN…
-
THAI LƯU SỚM
BS PHAN DIỄM ĐOAN NGỌC - BV TỪ DŨ Hồi…
-
NHÂN XƠ TỬ CUNG
BS.Phạm Thanh Hoàng- Giảng viên ĐHYD, BV Từ Dũ NHÂN…
-
SANH GIÚP
BS Đoàn Trung Hiếu - BV Từ Dũ Bài viết…
-
Nhiễm trùng vết mổ lấy thai
BS Nguyễn Phương Nam - BV Hùng Vương Nhân dịp…
-
NGHÉN (THAI HÀNH)
THS.BS.Phan Diễm Đoan Ngọc NGHÉN là tình trạng rất phổ…
-
ĐẾM CỬ ĐỘNG THAI
THS.BS. Phan Diễm Đoan Ngọc CỬ ĐỘNG THAI LÀ GÌ?…
-
MANG THAI VÀ CHUYỆN TIÊM NGỪA
BSCK1. Nguyễn Phương Nam – Bệnh viện Hùng Vương. Phụ…
-
THAI NHỎ, NHẸ CÂN – NỖI LO CỦA MẸ
BS CK1. NGUYỄN PHƯƠNG NAM – BỆNH VIỆN HÙNG VƯƠNG.…
-
TIỂU ĐƯỜNG (ĐÁI THÁO ĐƯỜNG) THAI KỲ
Bs. Phạm Thanh Hoàng Giảng viên đại học Y Dược…
-
SANH KHÔNG ĐAU
BS. Phạm Thanh Hoàng Giảng viên ĐHYD TPHCM - BV…
-
CẤY QUE NGỪA THAI
ThS.BS. Phan Diễm Đoan Ngọc Chào bạn, chọn biện pháp…
-
VÒNG TRÁNH THAI
VÒNG TRÁNH THAI (DỤNG CỤ TỬ CUNG) ThS.BS. Phan…
-
BIẾN ĐỔI LÀNH TÍNH CỔ TỬ CUNG
BIẾN ĐỔI LÀNH TÍNH CỔ TỬ CUNG: LỘ TUYẾN CỔ…
-
MẤT NGỦ KHI MANG THAI
BS. TRẦN THỊ MINH CHÂU. Mất ngủ khi mang thai…
-
PHỤ NỮ NÊN ĂN GÌ ĐỂ DỄ CÓ THAI?
BS. TRẦN THỊ MINH CHÂU ❓Bạn có biết: Khả năng…
-
DÙNG MỸ PHẨM TRONG THAI KỲ
ThS.BS. Phan Diễm Đoan Ngọc Nhân hôm nay có một…
-
DU LỊCH, ĐI LẠI TRONG THAI KỲ
BS.Phạm Thanh Hoàng Sắp đến Tết, nhiều người sẽ lên…
-
CANXI TRONG THAI KỲ
THS.BS.Phan Diễm Đoan Ngọc - BV Từ Dũ Hẳn bà…
-
LỢI ÍCH CỦA DẦU CÁ OMEGA-3 TRONG THAI KỲ
THS.BS.Phan Diễm Đoan Ngọc Trong thai kỳ, ngoài việc bổ…
-
THUỐC HỖ TRỢ PHỔI LÀ GÌ?
BS CK1. Nguyễn Phương Nam – Bệnh viện Hùng Vương.…
-
NÊN TIÊM NGỪA GÌ TRƯỚC KHI KẾT HÔN ?
BS. TRẦN THỊ MINH CHÂU Câu hỏi: Chào Bác sĩ,…
